







News
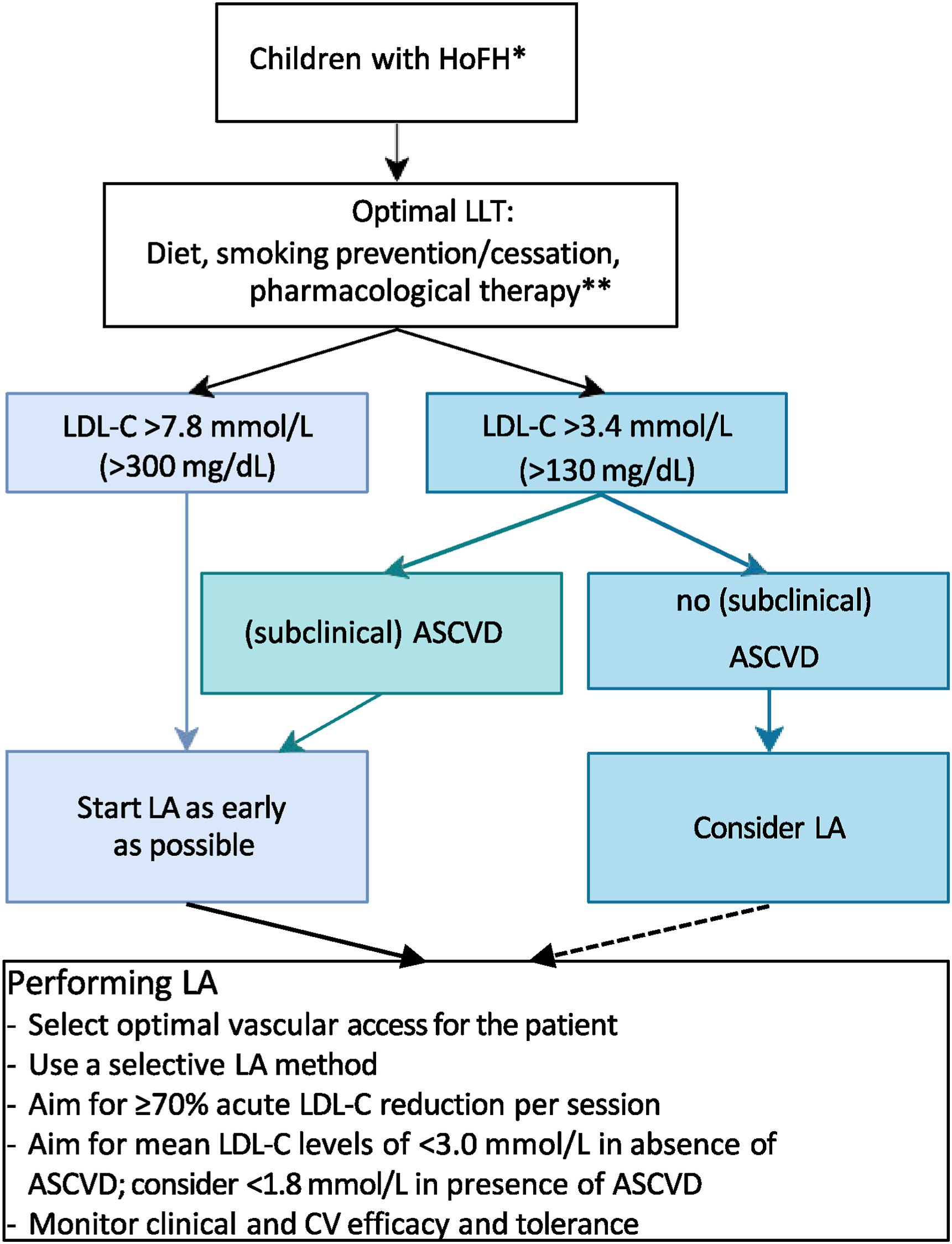
Just published in Atherosclerosis| CPR on lipoprotein apheresis for children with homozygous familial hypercholesterolaemia
An expert consensus statement from ERKNet and ESPN
Read more

Just published in Nature Reviews Nephrology! CPR for kidney involvement in tuberous sclerosis complex
A joint effort of ERKNet and ERA on providing a global overview on the management of TSC-associated kidney disease!
Read more

Follow ERKNet on social media!
Exciting times ahead as we unveil our redesigned social media presence tomorrow!
Read more
Events

ERKNet member: ERKReg Training Session
We regularly offer open trainings for new and advanced ERKReg editors. If you have new colleagues or need a refresher, register at least 3 days before…

CNE International Conference
The CNE international conference is hosted every second year and brings together scientists, clinicians, families and adult patients. Attendees from…
Follow us
On our social Networks